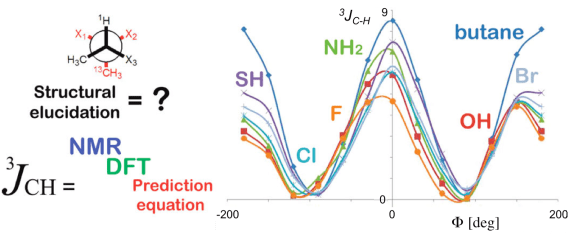
The 3J homo (1H-1H) and hetero-nuclear (1H-13C) NMR coupling constants are of fundamental importance for the structural elucidation of bio-organic & pharmaceutically relevant compounds. Since the correct interpretation of the NMR spectra for extrapolating the exact 3J values is a severe issue, quantum mechanical (QM) calculations are of great support for the prediction of the experimental data. An alternative approach is represented by the use of prediction equations, which are based on the first Karplus model (eq. 1).
 As a master student, I have developed a general hetero-nuclear 3JC-H prediction equation (eq. 3), based on the first homo-nuclear 3JH-H Karplus (eq.1) & Altona (eq. 2) equations (Palermo et al., J. Org. Chem. 2010). In agreement withe these models, our equation takes into account the electronegativity effect of the substituents attached to the 13C-C-C-1H fragment and the dihedral (Φ) dependence of the hetero-nuclear spin-coupling. Eq. 3 has been empirically derived, based on a dataset of ~2100 DFT MPW1PW91/6-31g(d,p) calculations on small butane/pentane models.
As a master student, I have developed a general hetero-nuclear 3JC-H prediction equation (eq. 3), based on the first homo-nuclear 3JH-H Karplus (eq.1) & Altona (eq. 2) equations (Palermo et al., J. Org. Chem. 2010). In agreement withe these models, our equation takes into account the electronegativity effect of the substituents attached to the 13C-C-C-1H fragment and the dihedral (Φ) dependence of the hetero-nuclear spin-coupling. Eq. 3 has been empirically derived, based on a dataset of ~2100 DFT MPW1PW91/6-31g(d,p) calculations on small butane/pentane models. 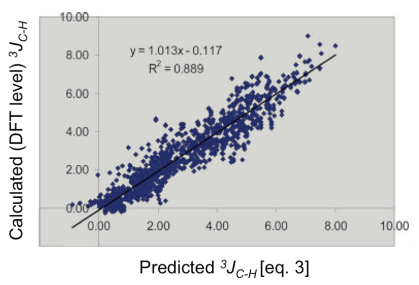
By plotting the calculated 3JC-H values (DFT level) versus the predicted 3JH-C (eq. 3), we found a linear distribution and a very high correlation (R3 = 0.889), which highlight the reliability of our model.
To confirm the accuracy of Eq. 3, we tested the equation on a large dataset of experimental 3JC-H. Low RMSD & average absolute difference (|ΔJ|/n) values demonstrate a good accuracy in the reproduction of the experimental NMR parameters. Several research group are now employing Eq. 3 as a support for the structural elucidation of the relative configuration of organic compounds.
Read more: J. Org. Chem., 2010, 75, pp 1982–1991
