Dynamics and Mechanism of CRISPR systems
We use computational methods to unravel the function and help the improvement of the CRISPR genome editing machineries.
Our multiscale approach is based on large-scale and ab-initio Molecular Dynamics, combined with cryo-EM processing approaches, free energy methods and network models derived on graph theory. This synergistic approach is used to determine the catalysis, allostery and specificity of CRISPR systems.
Publications
[1] Casalino et al. Accepted ACS Catalysis 2020. [2] Saha et al. Accepted J. Chem. Inf. Model. 2020. [3] Nierzwicki et al. WIREs Comp Mol Sci Accepted 2020. [4] East K. et al. J. Am. Chem. Soc. 2020. [5] Palermo G. J. Chem. Inf. Model. 2019.[6] Ricci C. G. et al.ACS Cent. Sci. 2019.[7] Palermo G. Chem 2019.[8] Palermo G. et al.Q. Rev. Biophys. 2018.[9] Palermo G. et al. J. Am. Chem. Soc. 2017. [10] Palermo G. et al. Proc. Natl. Acad. Sci. USA, 2017. [11] Palermo G. et al. ACS Cent. Sci. 2016.
Read about the invisible dance of CRISPR-Cas9 in Physics Today!
Allostery in Protein/Nucleic Acid Complexes

Many interesting allosteric targets are large protein/nucleic acid complexes that require facile communication between multi-domain structures for proper functionality. In these systems, structural remodeling and long-range allosteric communication are the main mechanistic determinants underlying their function in the nucleus of cells.
Our research combines Molecular Dynamics with network theory and centrality analysis to trace the allosteric signaling in large protein/nucleic acid complexes. By using this approach, we revealed a mechanism of allosteric drug-drug synergy in the Nucleosome Core Particle (Nat. Commun. 2017). Building on this early work, we found evidence of an intriguing allosteric mechanism in CRISPR-Cas9 (J. Am. Chem. Soc. 2020, J. Am. Chem. Soc. 2017, WIREs Comp. Mol. Sci. 2020).
Publications
[1] Nierzwicki et al. WIREs Comp. Mol. Sci. Accepted 2020. [2] East K. et al. J. Am. Chem. Soc. 2020. [3] Palermo G. et al. J. Am. Chem. Soc. 2017. [4] Adhrikenesan Z. et al. Nat. Commun. 2017.
Nucleosome Dynamics
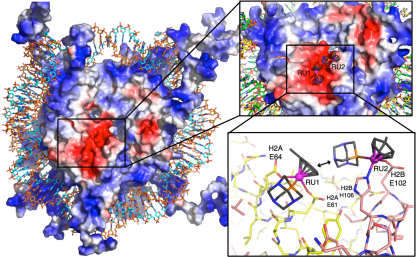
The Nucleosome Core Particle is a key constituent of chromatin and a molecular target for anticancer drugs. By integrating molecular dynamics with correlation models and graph theory, we have characterized the mechanism of action of metal-based anticancer agents at the level of the nucleosome core particle, which are promising as anticancer drugs.
Publications
[1] Adhrikenesan Z. et al. Nat. Commun. 2017. [2] Ma Z. et al. Angew. Chem. Int. Ed. 2016 [3] Palermo G. et al. ChemMedChem 2016
Membrane Proteins Structure and Dynamics

The Fatty Acid Amide Hydrolase is a key membrane protein involved in the control of pain. By using molecular dynamics techniques, including free energy methods and ab-initio MD, flanked by estensive Bayesian statistics analysis, we clarified the mechanisms of lipid selection and degradation in the enzyme, with insights for the discovery of targeting drugs.
Publications
[1] Vanni S. et al. Acc. Chem. Res. 2019. [2] Palermo G. et al. Eur. J. Med. Chem. 2015. [3] Palermo G. et al. J. Phys. Chem. B. 2015. [4] Palermo G. et al. J. Chem. Theory. Comput. 2013. [5] Palermo G. et al. J. Med. Chem. 2011.
Density Functional Theory for Solar Cells technology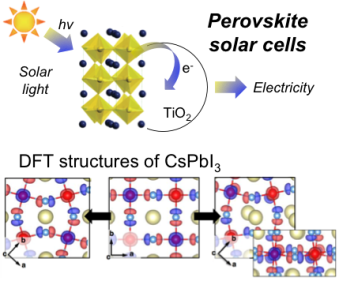
Solar cells are the energy revolution of the 21th century, converting solar energy in electricity. By using Density Functional Theory, we characterized the atomistic and electronic structure nature of hybrid organic-inorganic perovskites, novel materials for solar cells technology. Our outcomes have been integrated with the experiments of the lab of Prof. M. Graetzel for developing more efficient solar cells technologies.
Publications
[1] Meloni S. et al. J. Phys. Chem. C. 2017. [2] Ashari-Astani N. J. Mater. Chem. A 2017. [3] Meloni S. et al. http://arxiv.org/abs/1412.3659
A duel with pain: multi-target drug discovery
 Multi-target drug discovery is promising for the development of innovative drugs. By applying molecular simulations and free energy methods, I have clarified the mechanism of action of ARN2508, a novel anti-inflammatory agent that inhibits both the Fatty Acid Amide Hydrolase (FAAH) and the cyclooxygenase (COX) enzymes. With this research, we provide the basis of dual inhibition for anti-inflammatory treatments.
Multi-target drug discovery is promising for the development of innovative drugs. By applying molecular simulations and free energy methods, I have clarified the mechanism of action of ARN2508, a novel anti-inflammatory agent that inhibits both the Fatty Acid Amide Hydrolase (FAAH) and the cyclooxygenase (COX) enzymes. With this research, we provide the basis of dual inhibition for anti-inflammatory treatments.
Publications: Palermo G. et al. ChemMedChem 2016.
Structural elucidation of organic compounds
 My initial work has been focused on the development of methods for the prediction – based on the Karplus and Altona models – of NMR coupling constants (3JC-H), which are of key importance for the structural elucidation of bioorganic and pharmaceutically relevant compounds. Based on Density Functional Theory (DFT), I have formally derived a general 3JC-H prediction equation, which is used as a support to NMR experiments for the structural elucidation of organic compounds.
My initial work has been focused on the development of methods for the prediction – based on the Karplus and Altona models – of NMR coupling constants (3JC-H), which are of key importance for the structural elucidation of bioorganic and pharmaceutically relevant compounds. Based on Density Functional Theory (DFT), I have formally derived a general 3JC-H prediction equation, which is used as a support to NMR experiments for the structural elucidation of organic compounds.
Publications: Palermo G. et al. J. Org. Chem. 2010.
